A vital part of mathematical modelling is the accurate and reliable estimation of the model parameters. In biology, the required parameters are particularly difficult to measure because of either weaknesses in the measurement technology or a lack of direct measurements. In the latter case, parameters must be estimated from indirect measurements, usually in the form of time-series data, which are themselves sparse and noisy. The objective of this project is to address this problem by developing novel estimation methods that are particularly tailored to biological models consisting of nonlinear ordinary differential equations. Approaches taken in this project assume specific types of nonlinearities, such as generalized mass action, Michaelis Menten and Hill kinetics, and then use a suitable model extension that decouples the estimation of non-measured states from the parameters. This allows a two-step approach: (1) reconstruction of all extended states; (2) estimation of the parameters using these reconstructed states. An important advantage of the proposed method is that it allows us to identify suitable measurements and/or model structures for which the parameters can be estimated. In addition, it is generally applicable to models of metabolic networks, signal transduction and gene regulation.
Current Project:
Parameter estimation in biochemical reaction networks
Former Projects:
Multiscale Modeling of Neuronal Plasticity
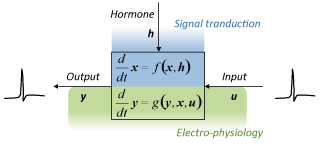
Hormones influence the intracellular state. This so called signal transduction is often modeld with (complex) reaction kinetic models deduced from the reaction graph (first principle model). The frequency of the eletrical impulses generated by the neuron depends on the synaptic input and can mathematically be described using Hodgin-Huxley models (simplified empirical descriptions of the ion-channels). The two domains are for example interconnected through the phosphorylation of ion channels. Integrated modeling of both domains is neccesary to understand how hormonic and synaptic signals are integrated.
The fundamental process underlying all brain function is the ability of the neurons to adapt to external inputs in the context of the neuronal state. The adaptive processes span multiple spatial and temporal scales ranging from millisecond dynamics of the ion channels, seconds to minutes time scale of the signalling pathways, and tens of minutes to hours time scale of the gene regulation and its feedback onto the signalling pathways and electro-physiology.
With the focus on the immediate AT1 receptor signalling dynamics and the consequences on the firing behaviour, we used mathematical modelling and analysis to decipher how the signals are integrated in this complex multi-scale system.
Work based at the Daniel Baugh Institute of functional genomics and computational biology at the Thomas Jefferson University in
| [1] | Rajanikanth Vadigepalli, Dirk Fey, and James S. Schwaber. Modeling neuronal adaptation in the brain: Integrating receptor signaling and electrophysiology. In FOSBE, 2007. (pdf 532kb) |
Domain oriented modeling of signal transduction
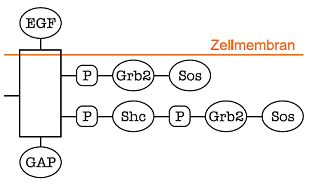
A EGF receptor with one extracellular and three intracellular binding sites, each able accumulate different signalling molecules. The number of possible monomer-states (different complex formations of the receptor monomer) is calculated to 22*41*61 = 96, resulting in 4560 dimer-states. In principle one has to consider all the different states, resulting in unsuitable lage models. If idependencies between the binding sites can be assumed, it is possible to reduce the model dimension dramatically.
In signal transduction, the formation of multi-protein complexes results in an unsuitable large models including a huge number of different species. For example, a transmembrane receptor is often subject of phosphorylation and multiple signalling protein assembly on several binding domains. Thereby the resulting number of species that are to include in the model (model dimension) increases exponentially with the number binding-sites and -proteins.
The model reduction method is based on a-priory knowledge on domain interactions, and reduces the model dimension dramatically (linear increase) in the case of independent domains. Extending this earlier approach, the application on a receptor dimerization revealed the systems inherent structure of its information flow.
Work based at the Institute for System Dynamics (ISys) at the University Stuttgart.